QUANTUM ESPRESSO:nscf
采用scf计算得到电荷密度和波函数进行NSCF计算, 非自洽(nscf)-采用更加密的K点采样及利用scf基态电荷密度计算,获得DOS(PDOS),Efermi;
设置nscf.in文件,需要修改的参数包括:calculation,nbnd,K_POINTS
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
&CONTROL
calculation = "nscf"
restart_mode = "from_scratch"
prefix = "carbyne"
outdir = "./outdir/"
pseudo_dir = "./pseudo/"
verbosity = "high"
tprnfor = .true.
tstress = .true.
etot_conv_thr = 1.0d-6
/
&SYSTEM
ibrav = 6
celldm(1) = 18.89726125
celldm(3) = 0.256551169
nat = 2
ntyp = 1
nbnd = 22
occupations = 'fixed'
ecutwfc = 50
ecutrho = 400
/
&ELECTRONS
conv_thr = 1.000e-9
electron_maxstep = 200
mixing_beta = 0.7
startingpot = "atomic"
startingwfc = "atomic+random"
/
K_POINTS {automatic}
1 1 400 0 0 0
ATOMIC_SPECIES
C 12.01070 C.pbe-n-kjpaw_psl.1.0.0.UPF
ATOMIC_POSITIONS (angstrom)
C 5.0000000000 5.0000000000 0.0000000000 0 0 0
C 5.0000000000 5.0000000000 1.2640744811 0 0 1
NSCF计算完成后,使用dos.x程序来处理得到态密度,输入文件dos.in如下
1
2
3
4
5
6
7
8
9
&dos
prefix = 'carbyne'
outdir = './outdir'
!bz_sum = 'tetrahedra_opt'
ngauss = 1
degauss = 0.005
deltaE = 0.01
fildos = 'carbyne.dos'
/
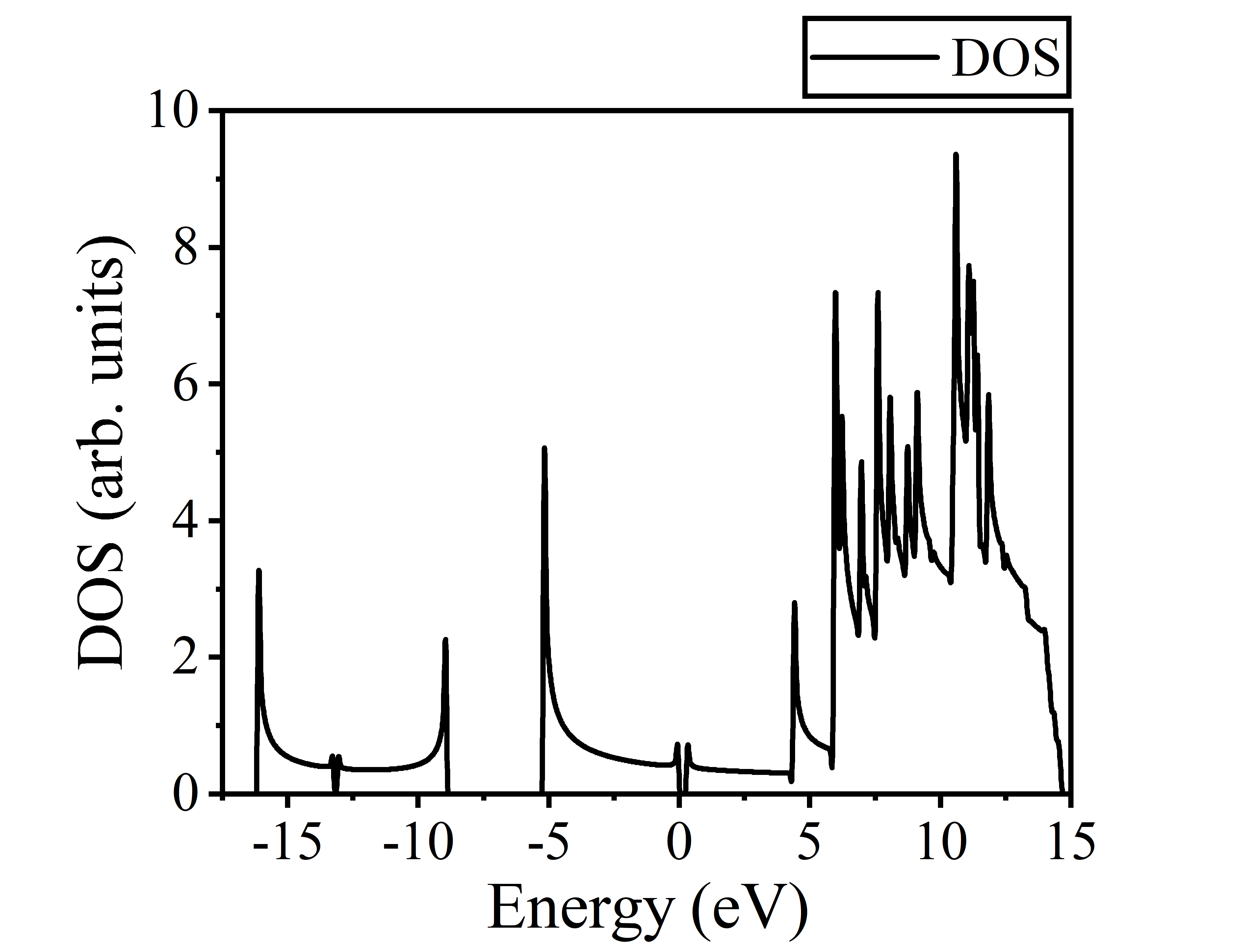
对文件carbyne.dos使用Originlab作图,并将能量减去VBM值,使得VBM=0 ,能态密度图如下:
使用projwfc.x计算投影态密度(PDOS)
Note: (在模守恒赝势下,不能得到投影态密度,[projwfc.x]会输出:“Cannot project on zero atomic wavefunctions”)
根据评论区Tang的意见,这个问题可以通过赝势生成软件ONCVPSP http://www.mat-simresearch.com 重新生成赝势解决,在此向Tang的建议表示感谢。
输入文件projwfc.in如下:
1
2
3
4
5
6
7
8
9
&projwfc
prefix = 'carbyne'
outdir = './outdir'
ngauss = 1
degauss = 5.0d-3
deltaE = 0.01
filpdos = 'carbyne.pdos'
filproj = 'carbyne.proj'
/
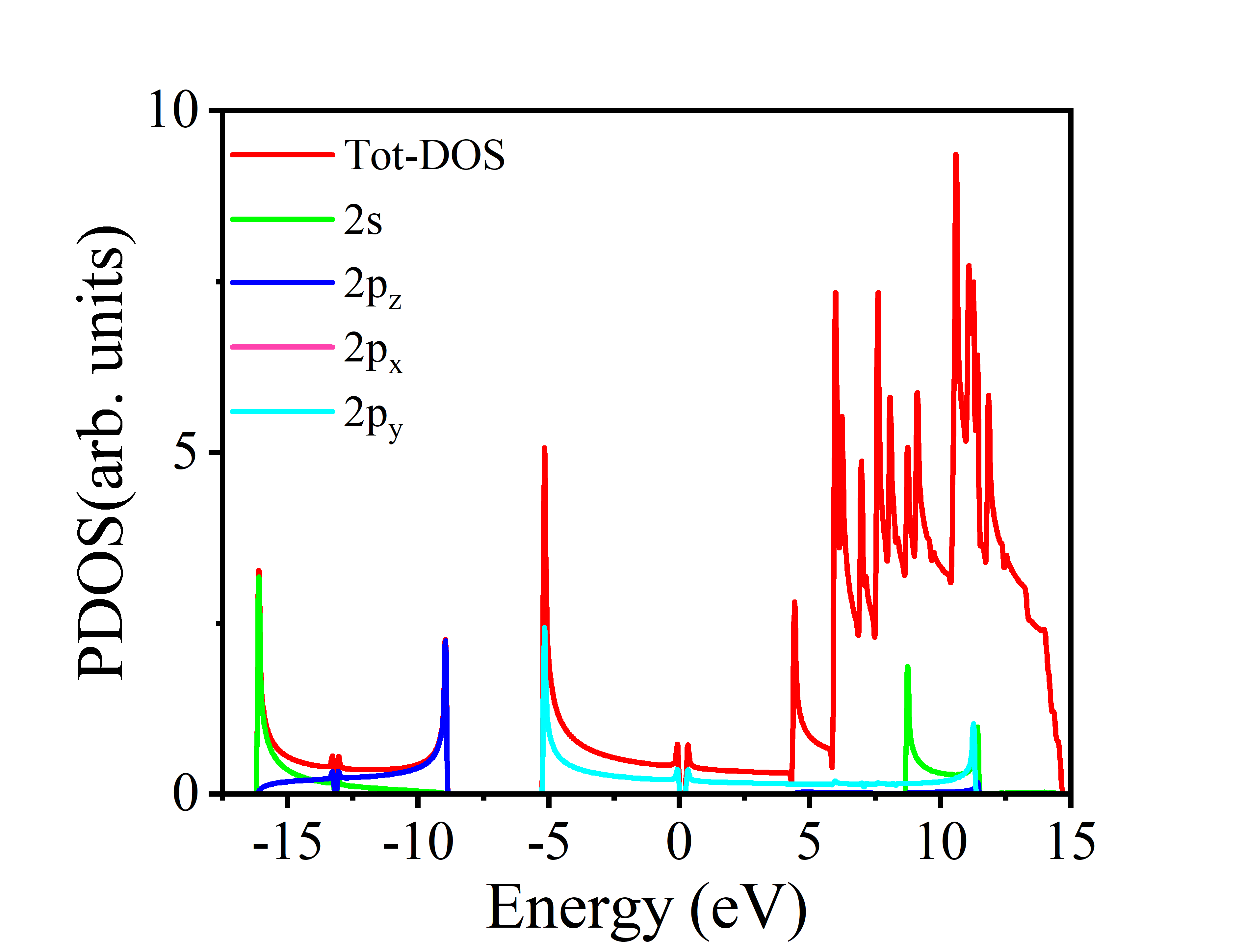
得到分波态密度图(PDOS)如下:
Note: 如遇到问题,欢迎在评论区留言。评论系统采用了Disqus系统,需要翻墙才能加载。