QUANTUM ESPRESSO:scf
自洽(scf)计算-获得体系基态电荷密度+波函数+构型能量
根据优化后结构的晶格常数和原子位置,设置scf.in文件
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
&CONTROL
calculation = "scf"
restart_mode = "from_scratch"
prefix = "carbyne"
outdir = "./outdir/"
pseudo_dir = "./pseudo/"
verbosity = "high"
tprnfor = .true.
tstress = .true.
etot_conv_thr = 1.0d-6
/
&SYSTEM
ibrav = 6
celldm(1) = 18.89726125
celldm(3) = 0.256551169
nat = 2
ntyp = 1
nbnd = 22
occupations = 'fixed'
ecutwfc = 50
ecutrho = 400
/
&ELECTRONS
conv_thr = 1.000e-9
electron_maxstep = 200
mixing_beta = 0.7
startingpot = "atomic"
startingwfc = "atomic+random"
/
K_POINTS {automatic}
1 1 200 0 0 0
ATOMIC_SPECIES
C 12.01070 C.pbe-n-kjpaw_psl.1.0.0.UPF
ATOMIC_POSITIONS (angstrom)
C 5.0000000000 5.0000000000 0.0000000000 0 0 0
C 5.0000000000 5.0000000000 1.2640744811 0 0 1
计算后得到的输出文件在文件夹outdir中,其中包括
- prefix.xml
- prefix.save
charge-density.dat (电荷密度文件)
*.UPF(赝势文件)
data-file-schema.xml
paw.txt
wfc*.dat(不可约k点的波函数)
使用下列命令可以得到体系的总能量:
1
grep ! scf.out
得到
1
! total energy = -36.74406518 Ry
使用pp.x处理得到one-electron (Kohn-Sham) orbitals或者分子轨道
输入文件psi2.in如下:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
&inputpp
prefix = 'carbyne'
outdir = './outdir'
filplot= 'psi2.carbyne'
plot_num = 7
kpoint = 101
kband(1) = 1
kband(2) = 8
!lsign = .true.
/
&plot
fileout = '.xsf'
iflag = 3
nfile = 1
output_format = 5
weight(1)= 1.0
/
输入文件具体参数设置,参考QE安装包“qe-6.8/PP/Doc/INPUT_PP.html”中的说明
其中lsign=.true. if true and k point is Gamma, plot |psi|^2 sign(psi)
输出文件得到:filplot_K_B.xsf,可以使用VESTA作图,显示波函数。
VBM以及CBM在布里渊区边界,且为二重简并,电荷密度如下图:
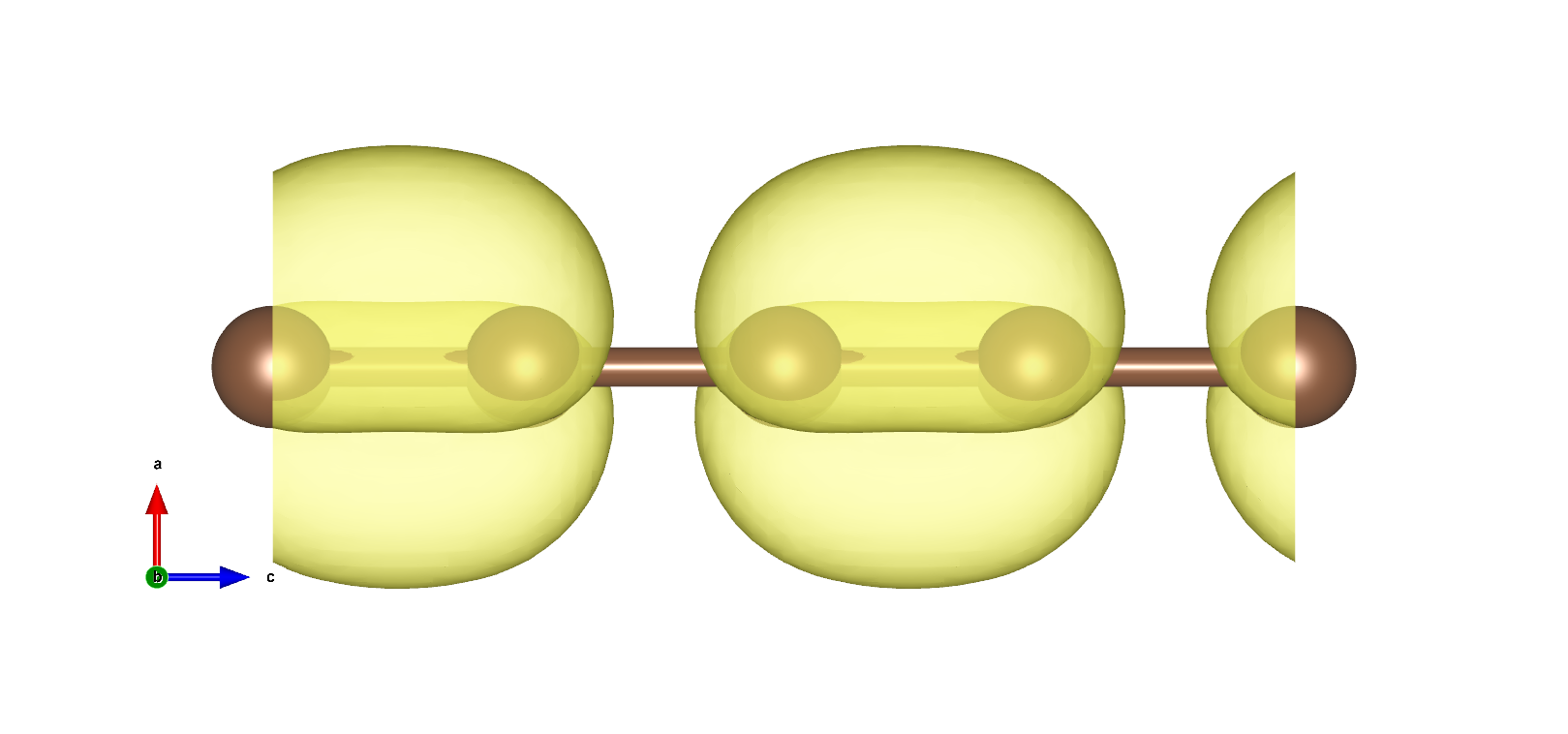
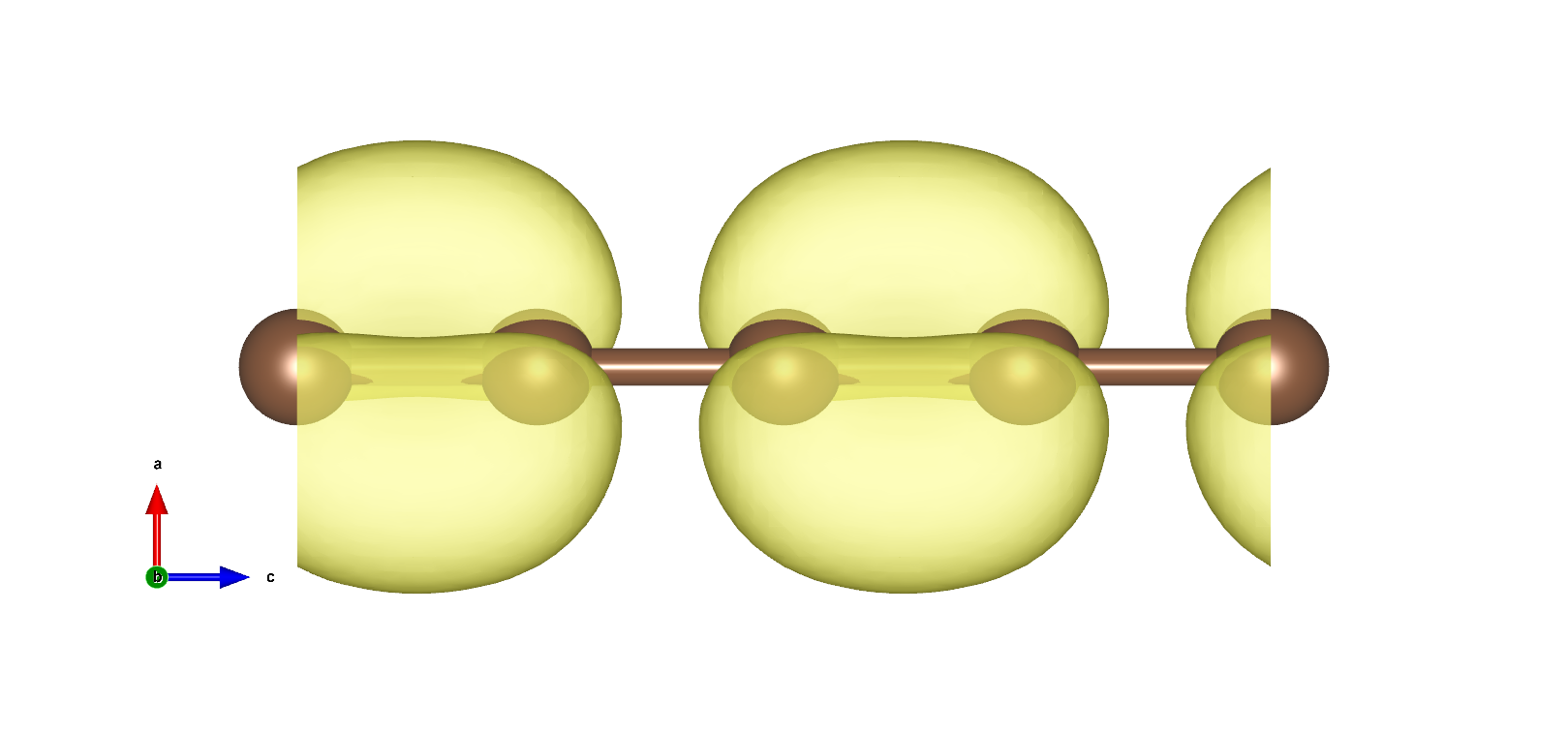
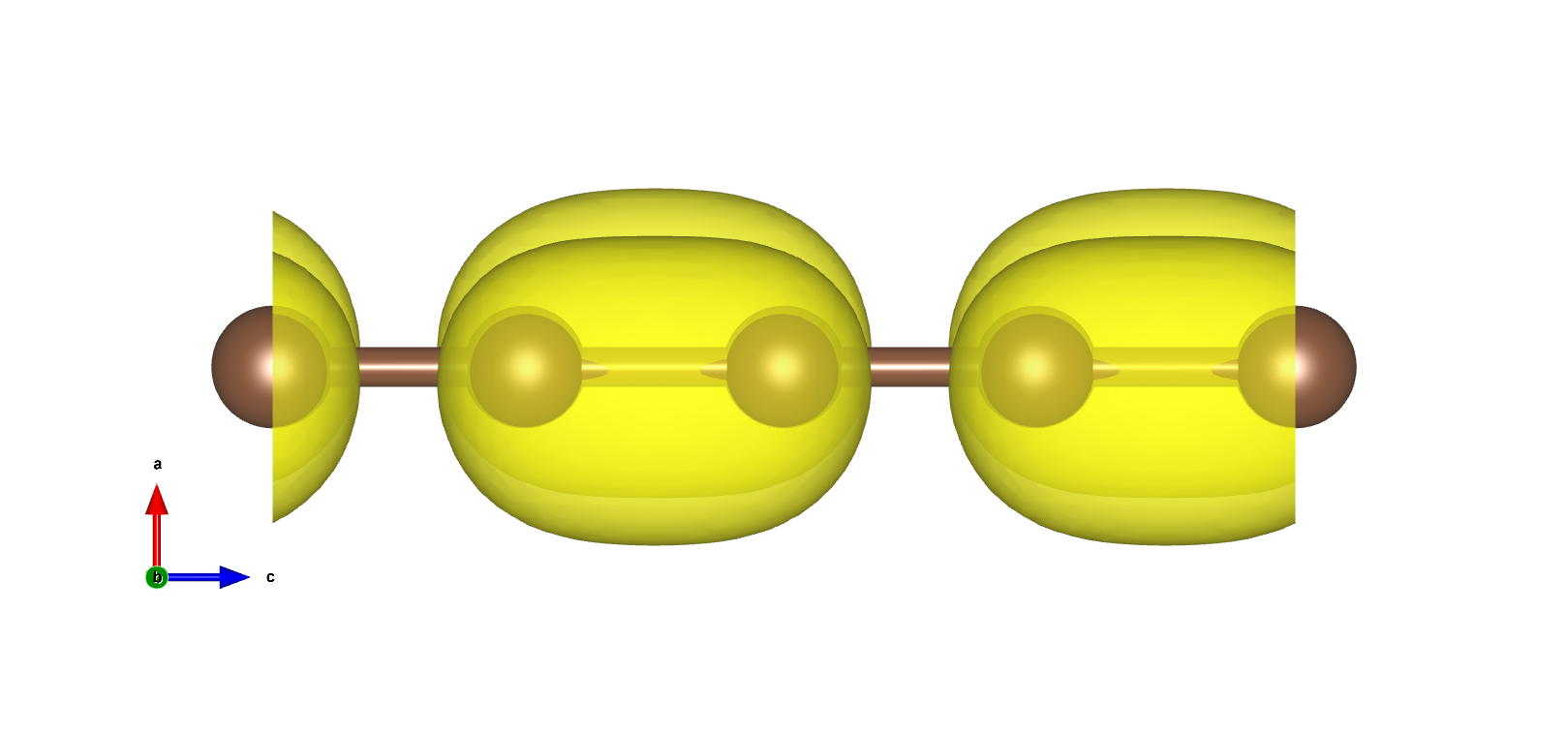
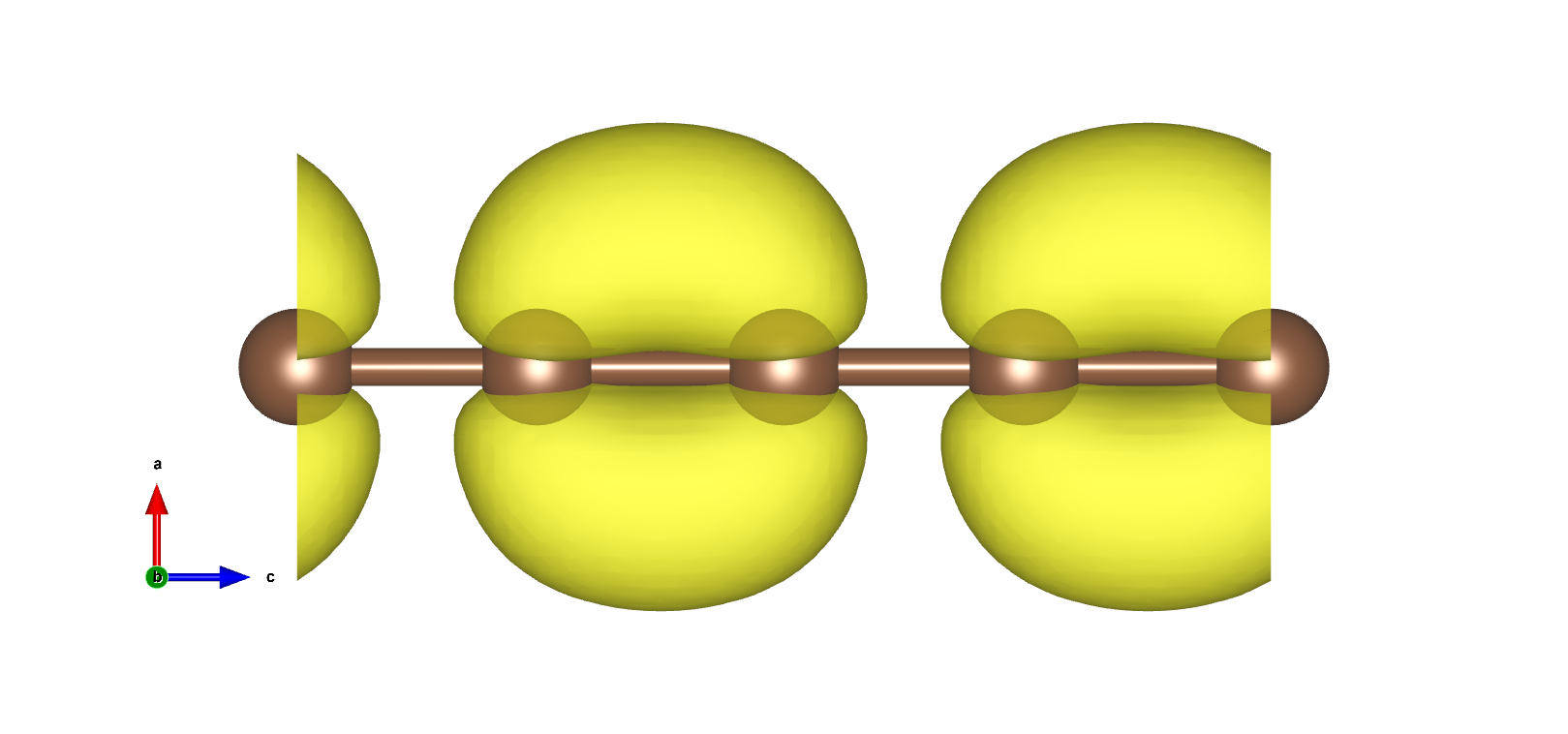
使用pp.x处理得到电荷密度图
输入文件rho.in如下:
1
2
3
4
5
6
7
8
9
10
11
12
13
&inputpp
prefix = 'carbyne'
outdir = './outdir'
filplot= 'carbyne.rho'
plot_num = 0
/
&plot
filepp(1)= 'carbyne.rho'
iflag = 3
output_format = 5
fileout = 'carbyne.rho.xsf'
/
使用VESTA打开得到3D电荷密度图如下所示:
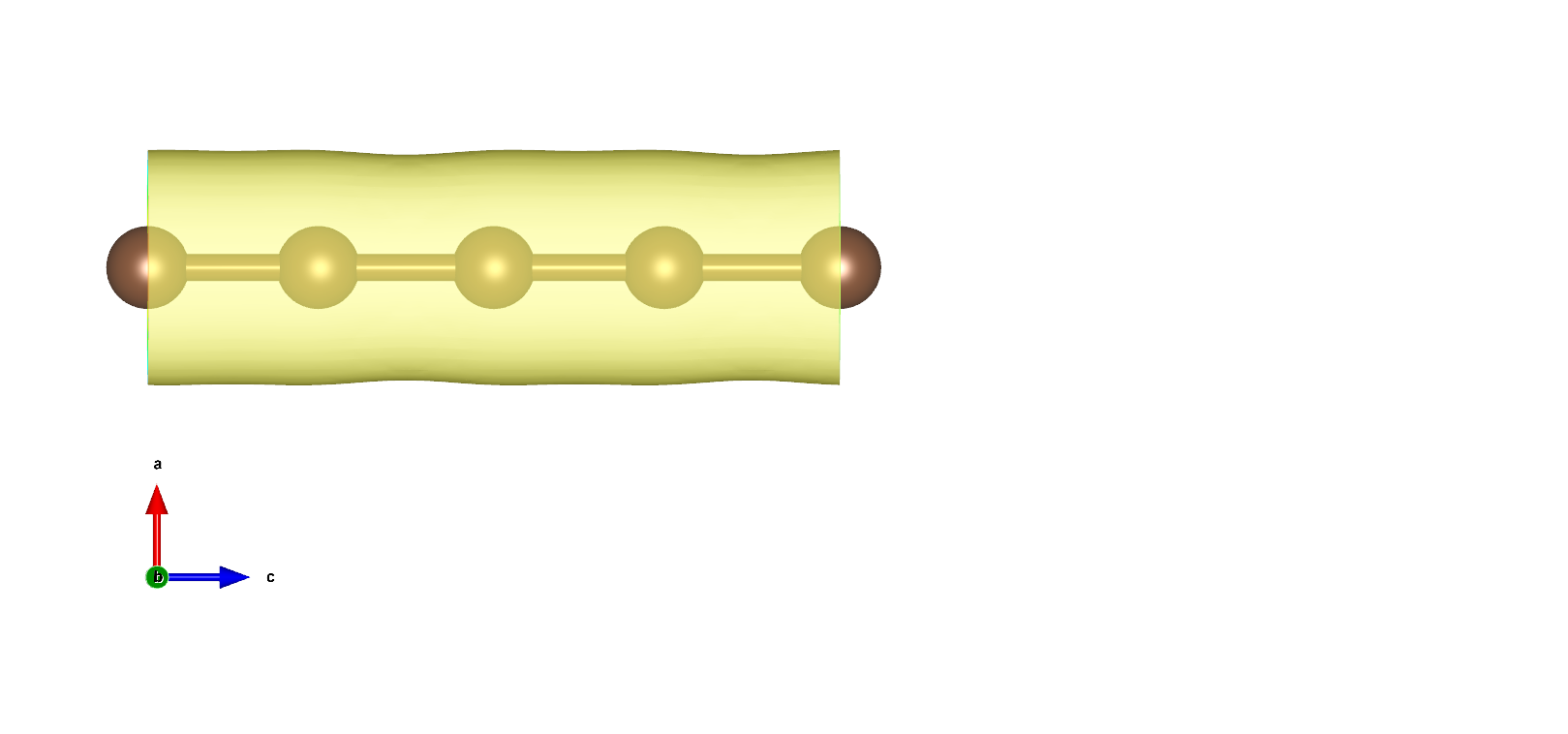
在VESTA中设置Boundary,将z(max)设置显示跟多单胞,设置Cutoff planes得到电荷密度截面:
