QUANTUM ESPRESSO:ph.x计算phonon
使用DFPT计算声子色散谱,简正模分析,声子态密度,电声子耦合,红外和Raman光谱
Note:(1)为了后续epw.x的计算,在scf计算中设置outdir='./'
(2)phonon计算中设置outdir='./'、fildyn='prefix.dyn'以及fildvscf='dvscf'
计算步骤如下:
-
对relax后的结构,用pw.x运行‘scf’计算,对于ph.x计算,需要对结构优化和scf计算的收敛精度调高
pw.x <scf.in> scf.out -
使用
ph.x计算$\Gamma$点phonon或者phonon dispersion在多个q-points,其输入文件ph.in分别如下:Perform a phonon calculation at $\Gamma$ using the program
1 2 3 4 5 6 7 8 9 10 11 12 13
Phonons at Gamma &inputph tr2_ph=1.0d-16, prefix='carbyne', outdir='./' epsil=.true. !use for insulators and semiconductor with Gamma point !ldisp=.true. !nq1=1, nq2=1, nq3=5 !amass(1)=0.0 fildyn=“prefix.dyn”, fildvscf='dvscf' / 0.0 0.0 0.0
对于dispersion phonon计算,使用
ldisp=.true.和nq1=,nq2=,nq3=参数,取消文件末尾的q point
对于a polar semiconductor(极性半导体材料),需要设置epsil=.true.用来计算和存储介$\Gamma$点的宏观介电张量和Born有效电荷 -
对于采用
ph.x计算的出来的phonon频率,由于数字计算的不准确性,使得结果不满足$\Gamma$点的声学支求和为0的规则。并且对于非极性半导体材料,不满足LO-TO分裂。- 对计算$\Gamma$点phonon频率的,可以直接采用dynmat.x进行修正.
输入文件
dynmat.prefix.in如下1 2 3 4 5 6
&input fildyn = "prefix.dyn" asr = 'simple' q(1) = 1.0 q(2) = 0.0 q(3) = 0.0
其中
q(1),q(2),q(3)为direction for the LO-TO splitting运行
dynmat.x < dynmat.prefix.in>dynmat.prefix.outNOTE: 输出文件不要使用
dynmat.out,因为dynmat.x 的输入文件的filout 默认为dynmat.out- 对于phonon dispersion 计算,采用ph.x进行DFPT计算后。使用
q2r.x进行傅里叶变换得到实空间中的原子间力常矩阵
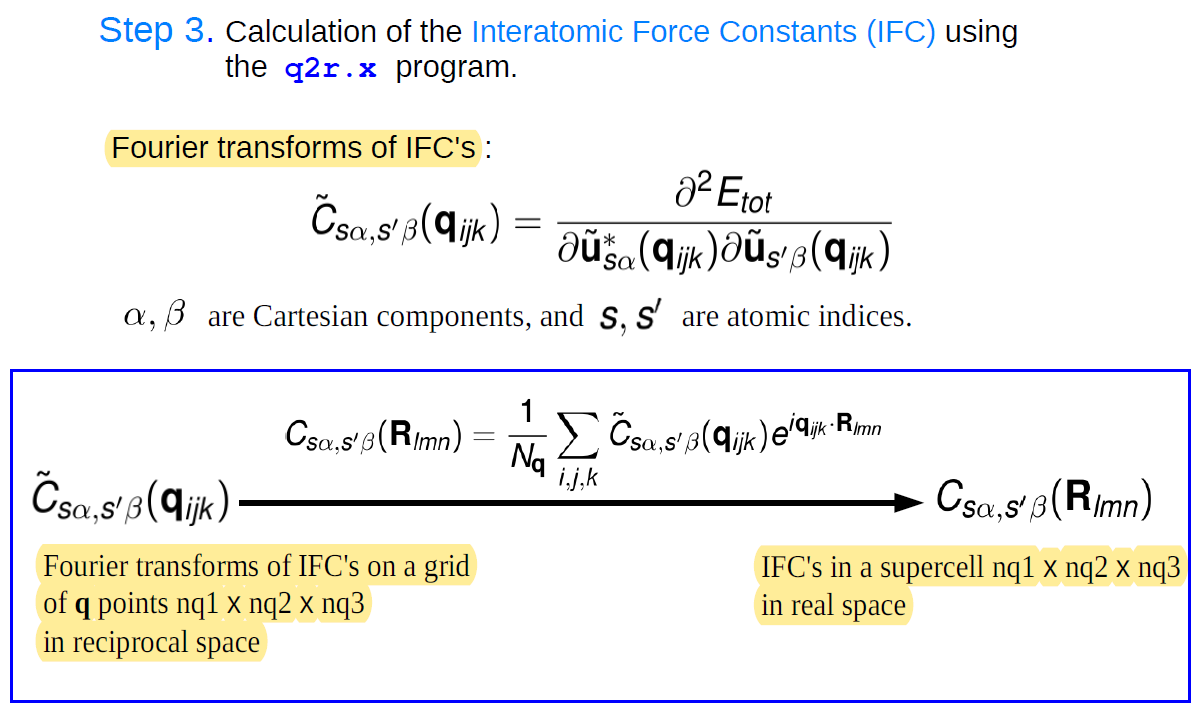
q2r.x的输入文件q2r.in如下:
1 2 3 4
&input fildyn='prefix.dyn' zasr='simple' flfrc='prefix.fc'
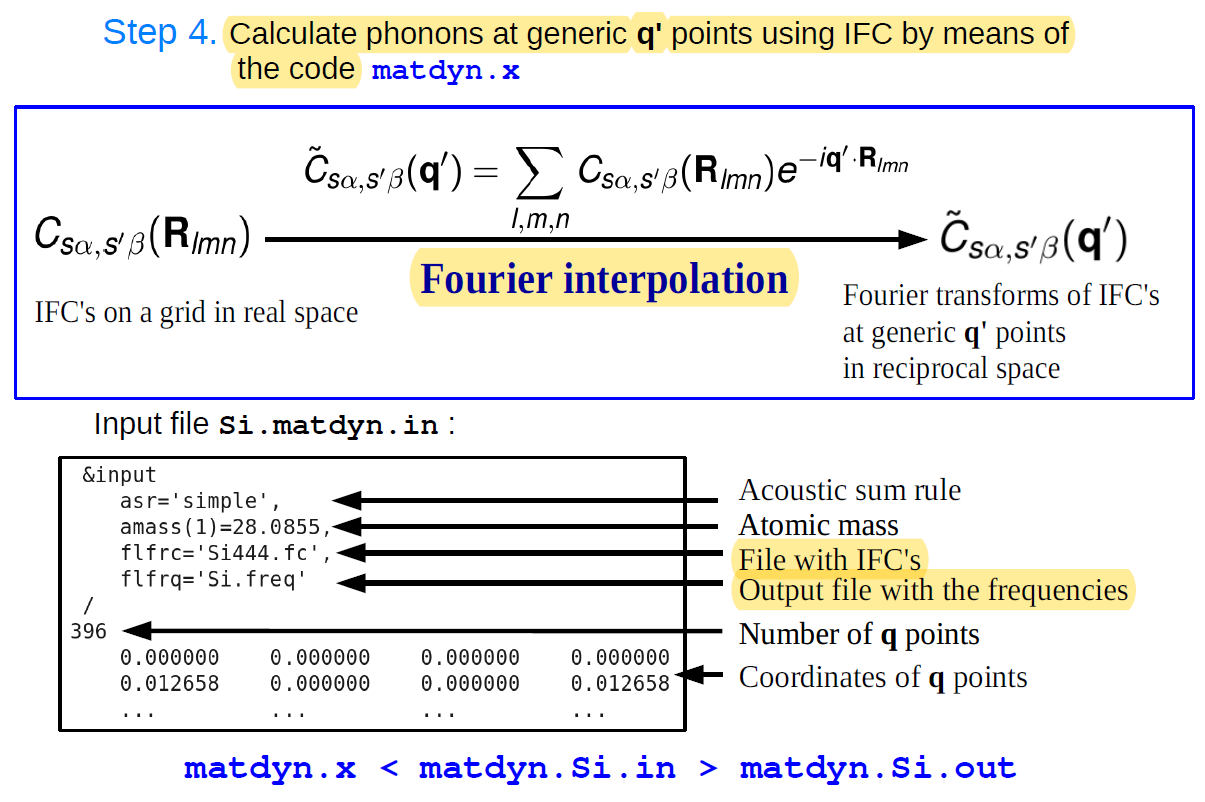
运行 q2r.x < q2r.in>q2r.out使用q2r.x计算出来的原子间力常数,则可以使用
dynmat.x计算出任意q点的声子频率以及声子态密度和声子色散谱。其输入文件如下:matdyn-dos.in
1 2 3 4 5 6 7 8
&input asr='simple', ! amass(1)=26.98, amass(2)=74.922, flfrc='flfrc', dos=.true. fldos="phonon.dos" nk1=1,nk2=1,nk3=200 /
matdyn-freq.in
1 2 3 4 5 6 7 8 9 10 11
&input asr='simple', ! amass(1)=26.98, amass(2)=74.922, flfrc='flfrc', flfrq='phonon-freq', q_in_band_form=.true., / 3 0.0 0.0 -0.5 100 0.0 0.0 0.0 100 0.0 0.0 0.5 1
使用
plotband.x来提取声子能带数据:1 2 3 4 5 6
phonon-freq 0 4000 phonon-freq.plot phonon-freq.ps 0.0 0.1 0.0
