QUANTUM ESPRESSO参数设置收敛性测试:
pw.x的参数在设置的时候,需要进行收敛性测试,以使得计算得到的结果满足收敛性条件,在满足计算精度的情况下尽量减少计算量,常见的需要进行收敛性测试的参数包括:平面波截断能ecutwfc、电荷密度和电势的截断能ecutrho(在使用超软赝势和PAW赝势时需要进行收敛性测试)、k-point、SCF计算收敛精度conv_thr、对于金属进行布里渊区积分时的展宽系数degauss的收敛性测试。对于表面模型,还需要对表面模型的原子层数进行收敛性测试。对于收敛性测试,一般的收敛性判据为使得能量变化值小于1meV/atom(7.35$\times10^-5Ry/atom$ ),对于计算量较大的体系,可以适当降低计算精度。对于收敛性测试,一般需要先使用高精度的方法对晶胞进行结构优化,在优化好的结构基础上进行收敛性测试。使用状态方程(Equation of State)拟合得到晶格常数。
(1) ecutwfc的收敛性测试
收敛性测试脚本:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
#!/bin/bash
#BSUB -J ecuttest
#BSUB -q privateq-zw
##BSUB -q publicq
#BSUB -n 28
#BSUB -R "span[ptile=28]"
#BSUB -o %J.out
#BSUB -e %J.err
CURDIR=$PWD
rm -f nodelist >& /dev/null
for host in `echo $LSB_HOSTS`
do
echo $host >> nodelist
done
NP=`cat nodelist | wc -l`
rm nodelist
module use /share/home/zw/xiehua/modulefiles
module load Quantum_Espresso/7.0
rm -f etot_vs_ecut.dat
for ecut in $(seq 40 5 100)
do
erho=$[8 * $ecut]
cat >scf.$ecut.in<< EOF
&CONTROL
calculation = "scf"
restart_mode = "from_scratch"
prefix = "graphene"
outdir = "./"
pseudo_dir = "./pseudo/"
verbosity = "high"
tprnfor = .true.
tstress = .true.
etot_conv_thr = 1.0d-8
forc_conv_thr = 1.0d-7
/
&SYSTEM
ibrav = 4
nat = 2
ntyp = 1
! a = 2.464
! c = 15.0
celldm(1) = 4.66148920
celldm(3) = 6.08086614
! nbnd = 16
assume_isolated = "2D"
occupations = "smearing"
smearing = "mp"
degauss = 1.0d-2
ecutwfc = $ecut
ecutrho = $erho
/
&ELECTRONS
conv_thr = 1.000e-12
electron_maxstep = 200
mixing_beta = 7.00000e-01
startingpot = "atomic"
startingwfc = "atomic+random"
/
!&IONS
! ion_dynamics = "bfgs"
!/
!&CELL
! cell_dofree = "ibrav+2Dxy"
! cell_dynamics = "bfgs"
! press_conv_thr = 0.001
!/
K_POINTS {automatic}
36 36 1 0 0 0
ATOMIC_SPECIES
C 12.01070 C.pbe-n-kjpaw_psl.1.0.0.UPF
ATOMIC_POSITIONS {crystal}
C 0.333333333333333 0.666666666666667 0.500000
C 0.666666666666667 0.333333333333333 0.500000
EOF
mpirun -np $NP pw.x -nk 7 <scf.$ecut.in>scf.$ecut.out
grep -e 'kinetic-energy cutoff' -e ! scf.$ecut.out | \
awk '/kinetic-energy/ {ecut=$(NF-1)}
/!/ {print ecut, $(NF-1)}' >> etot_vs_ecut.dat
done
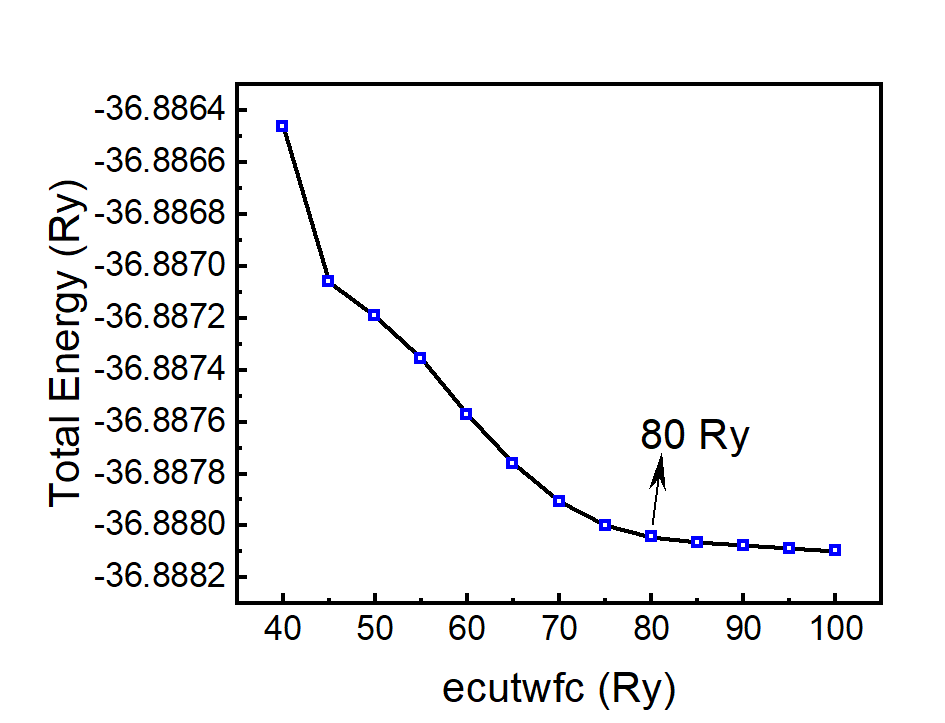
(2) 在得到平面波截断能的收敛能后测试电势的截断能ecutrho收敛性
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
#!/bin/bash
#BSUB -J ecuttest
#BSUB -q privateq-zw
##BSUB -q publicq
#BSUB -n 28
#BSUB -R "span[ptile=28]"
#BSUB -o %J.out
#BSUB -e %J.err
CURDIR=$PWD
rm -f nodelist >& /dev/null
for host in `echo $LSB_HOSTS`
do
echo $host >> nodelist
done
NP=`cat nodelist | wc -l`
rm nodelist
module use /share/home/zw/xiehua/modulefiles
module load Quantum_Espresso/7.0
rm -f etot_vs_ecut.dat
for rho in $(seq 4 1 16)
do
erho=$[80 * $rho]
cat >scf.$erho.in<< EOF
&CONTROL
calculation = "scf"
restart_mode = "from_scratch"
prefix = "graphene"
outdir = "./"
pseudo_dir = "./pseudo/"
verbosity = "high"
tprnfor = .true.
tstress = .true.
etot_conv_thr = 1.0d-8
forc_conv_thr = 1.0d-7
/
&SYSTEM
ibrav = 4
nat = 2
ntyp = 1
! a = 2.464
! c = 15.0
celldm(1) = 4.66148920
celldm(3) = 6.08086614
! nbnd = 16
assume_isolated = "2D"
occupations = "smearing"
smearing = "mp"
degauss = 1.0d-2
ecutwfc = 80
ecutrho = $erho
/
&ELECTRONS
conv_thr = 1.000e-12
electron_maxstep = 200
mixing_beta = 7.00000e-01
startingpot = "atomic"
startingwfc = "atomic+random"
/
!&IONS
! ion_dynamics = "bfgs"
!/
!&CELL
! cell_dofree = "ibrav+2Dxy"
! cell_dynamics = "bfgs"
! press_conv_thr = 0.001
!/
K_POINTS {automatic}
36 36 1 0 0 0
ATOMIC_SPECIES
C 12.01070 C.pbe-n-kjpaw_psl.1.0.0.UPF
ATOMIC_POSITIONS {crystal}
C 0.333333333333333 0.666666666666667 0.500000
C 0.666666666666667 0.333333333333333 0.500000
EOF
mpirun -np $NP pw.x -nk 7 <scf.$erho.in>scf.$erho.out
grep -e 'charge density cutoff' -e ! scf.$erho.out | \
awk '/charge density cutoff/ {ecut=$(NF-1)}
/!/ {print ecut, $(NF-1)}' >> etot_vs_erho.dat
done
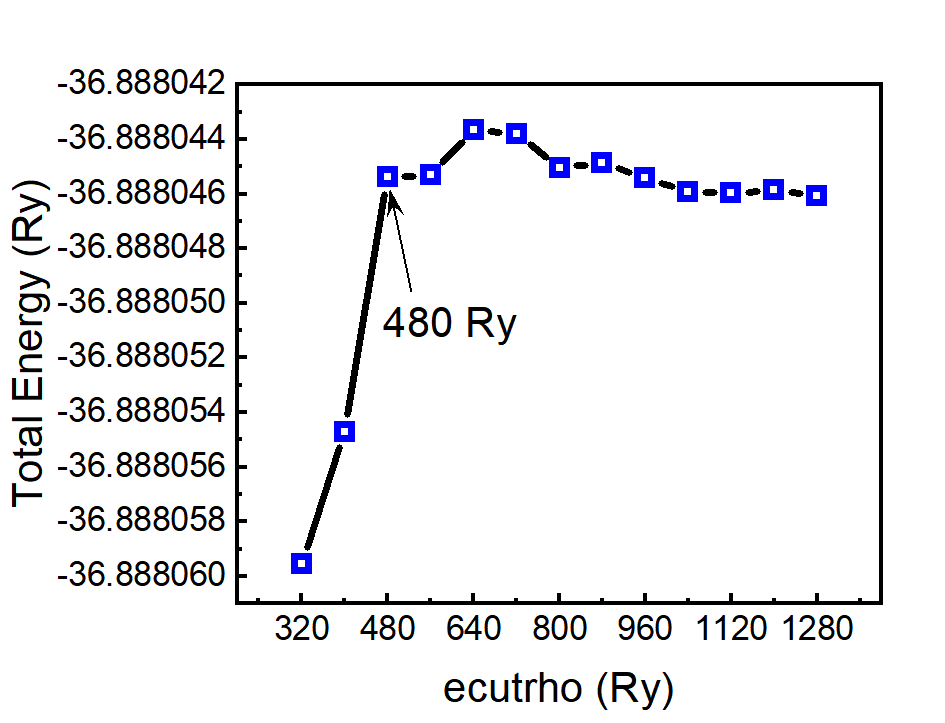
(3) 测试k_points的收敛性
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
#!/bin/bash
#BSUB -J kpointtest
#BSUB -q privateq-zw
##BSUB -q publicq
#BSUB -n 28
#BSUB -R "span[ptile=28]"
#BSUB -o %J.out
#BSUB -e %J.err
CURDIR=$PWD
rm -f nodelist >& /dev/null
for host in `echo $LSB_HOSTS`
do
echo $host >> nodelist
done
NP=`cat nodelist | wc -l`
rm nodelist
module use /share/home/zw/xiehua/modulefiles
module load Quantum_Espresso/7.0
rm -f etot_vs_nk.dat
for nk in $(seq 6 6 60)
do
cat >scf.nk$nk.in<< EOF
&CONTROL
calculation = "scf"
restart_mode = "from_scratch"
prefix = "graphene"
outdir = "./"
pseudo_dir = "./pseudo/"
verbosity = "high"
tprnfor = .true.
tstress = .true.
etot_conv_thr = 1.0d-8
forc_conv_thr = 1.0d-7
/
&SYSTEM
ibrav = 4
nat = 2
ntyp = 1
! a = 2.464
! c = 15.0
celldm(1) = 4.66148920
celldm(3) = 6.08086614
! nbnd = 16
assume_isolated = "2D"
occupations = "smearing"
smearing = "mp"
degauss = 1.0d-2
ecutwfc = 80
ecutrho = 480
/
&ELECTRONS
conv_thr = 1.000e-12
electron_maxstep = 200
mixing_beta = 7.00000e-01
startingpot = "atomic"
startingwfc = "atomic+random"
/
!&IONS
! ion_dynamics = "bfgs"
!/
!&CELL
! cell_dofree = "ibrav+2Dxy"
! cell_dynamics = "bfgs"
! press_conv_thr = 0.001
!/
K_POINTS {automatic}
$nk $nk 1 0 0 0
ATOMIC_SPECIES
C 12.01070 C.pbe-n-kjpaw_psl.1.0.0.UPF
ATOMIC_POSITIONS {crystal}
C 0.333333333333333 0.666666666666667 0.500000
C 0.666666666666667 0.333333333333333 0.500000
EOF
mpirun -np $NP pw.x -nk 7 <scf.nk$nk.in>scf.nk$nk.out
E=$(grep -e ! scf.nk$nk.out | awk '{print $(NF-1)}')
echo $nk $E >> etot_vs_nk.dat
done
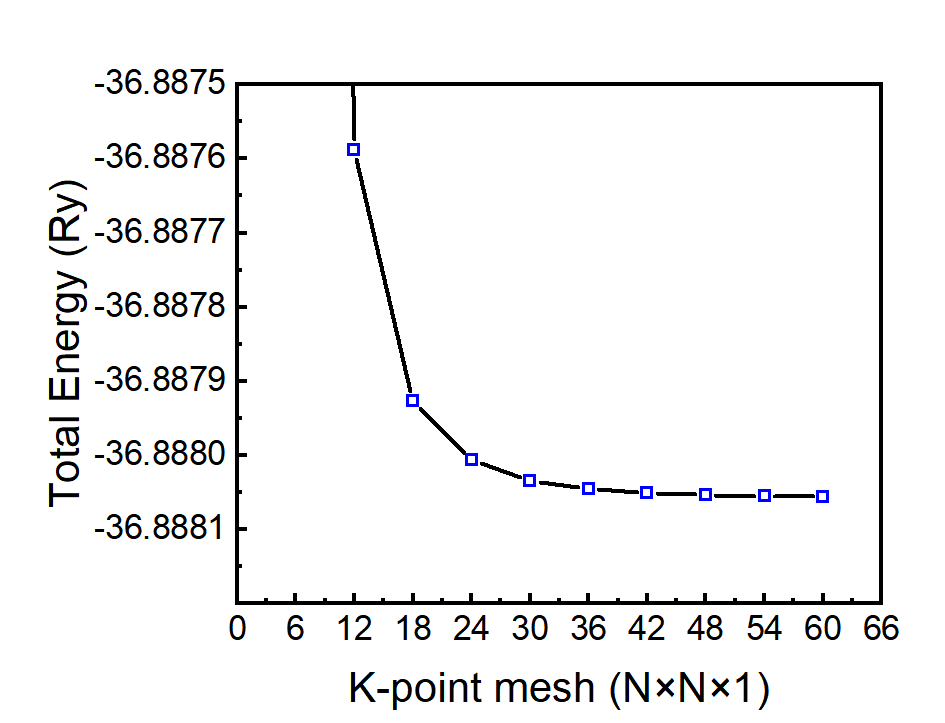
使用36×36×1,或者更密的k点采样。
(4) 使用不同的晶格常数进行scf计算,采用EOS方程拟合得到晶格常数
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
#!/bin/bash
#BSUB -J EoS
#BSUB -q privateq-zw
##BSUB -q publicq
#BSUB -n 28
#BSUB -R "span[ptile=28]"
#BSUB -o %J.out
#BSUB -e %J.err
CURDIR=$PWD
rm -f nodelist >& /dev/null
for host in `echo $LSB_HOSTS`
do
echo $host >> nodelist
done
NP=`cat nodelist | wc -l`
rm nodelist
module use /share/home/zw/xiehua/modulefiles
module load Quantum_Espresso/7.0
rm -f etot_vs_lattice.dat
for lattice in $(seq 2.455 0.002 2.475)
do
cat >scf.a$lattice.in<< EOF
&CONTROL
calculation = "scf"
restart_mode = "from_scratch"
prefix = "graphene"
outdir = "./"
pseudo_dir = "./pseudo/"
verbosity = "high"
tprnfor = .true.
tstress = .true.
etot_conv_thr = 1.0d-8
forc_conv_thr = 1.0d-7
/
&SYSTEM
ibrav = 4
nat = 2
ntyp = 1
a = $lattice
c = 20
! celldm(1) = 4.66148920
! celldm(3) = 6.08086614
! nbnd = 16
assume_isolated = "2D"
occupations = "smearing"
smearing = "mp"
degauss = 1.0d-2
ecutwfc = 80
ecutrho = 480
/
&ELECTRONS
conv_thr = 1.000e-12
electron_maxstep = 200
mixing_beta = 7.00000e-01
startingpot = "atomic"
startingwfc = "atomic+random"
/
!&IONS
! ion_dynamics = "bfgs"
!/
!&CELL
! cell_dofree = "ibrav+2Dxy"
! cell_dynamics = "bfgs"
! press_conv_thr = 0.001
!/
K_POINTS {automatic}
36 36 1 0 0 0
ATOMIC_SPECIES
C 12.01070 C.pbe-n-kjpaw_psl.1.0.0.UPF
ATOMIC_POSITIONS {crystal}
C 0.333333333333333 0.666666666666667 0.500000
C 0.666666666666667 0.333333333333333 0.500000
EOF
mpirun -np $NP pw.x -nk 7 <scf.a$lattice.in>scf.a$lattice.out
E=$(grep -e ! scf.a$lattice.out | awk '{print $(NF-1)}')
echo $lattice $E >> etot_vs_lattice.dat
done
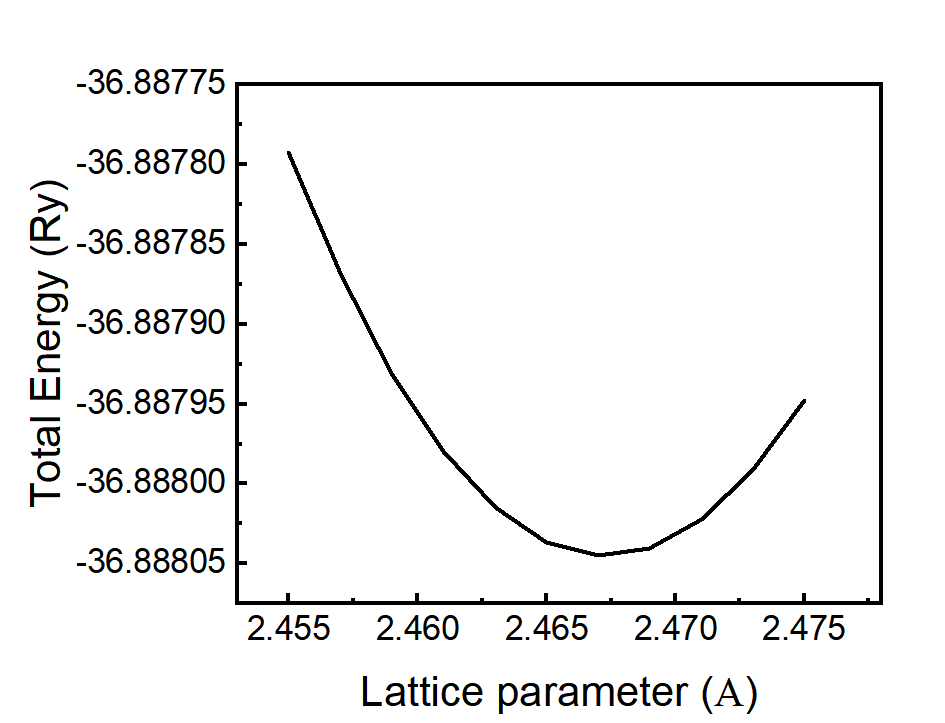
采用QE的ev.x拟合得到晶格常数a=2.46730242(对于QE中采用vc-relax进行晶格优化,经常导致原子偏离高对称点,尤其是对于超胞,建议先使用EOS方程进行拟合得到晶格常数,再优化)
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
# equation of state: murnaghan. chisq = 0.7005D-11
# V0 = 711.52 a.u.^3, k0 = 1038 kbar, dk0 = 9.00 d2k0 = 0.000 emin = -36.88805
# V0 = 105.44 Ang^3, k0 = 103.8 GPa
##########################################################################
# Vol. E_calc E_fit E_diff Pressure Enthalpy
# Ang^3 Ry Ry Ry GPa Ry
##########################################################################
104.39 -36.88779 -36.88779 0.00000 1.08 -36.83599
104.56 -36.88787 -36.88787 -0.00000 0.90 -36.84479
104.73 -36.88793 -36.88793 0.00000 0.72 -36.85347
104.90 -36.88798 -36.88798 -0.00000 0.54 -36.86203
105.07 -36.88801 -36.88802 0.00001 0.36 -36.87046
105.24 -36.88804 -36.88804 -0.00000 0.19 -36.87880
105.41 -36.88805 -36.88805 -0.00000 0.02 -36.88701
105.59 -36.88804 -36.88804 0.00000 -0.15 -36.89510
105.76 -36.88802 -36.88802 0.00000 -0.31 -36.90309
105.93 -36.88799 -36.88799 0.00000 -0.47 -36.91097
106.10 -36.88795 -36.88795 -0.00000 -0.63 -36.91874
$V0 = \sqrt3/2 \times a^2 \times c$ 则可以得到晶格常数$a=2.467302421337 Ang$